(This is an updated version of my earlier writing, which has just been published at GNEWS: https://gnews.org/192144/. Unfortunately, the GNEWS version has a few editing errors. So, I am posting the draft here as well. If you have read my earlier writing before, sorry for repeating some of the things here again. In that case, please skip the first section “who dares to carry out such a deceitful action”. Thank you!)
What is the true origin of the Wuhan coronavirus (SARS-CoV-2, 2019nCoV, the CCP virus)? Many scientific publications seem to tell you that the virus was born from nature. How reliable are these publications? While, before I comment on that, I would like to bring out an important fact: all of such publications rely on a single evidence – the sequence of a bat coronavirus named RaTG13.
RaTG13 looks like a “close cousin” of the Wuhan coronavirus – the two are 96% identical throughout the whole sequence of the viral genome. If RaTG13 is a nature-borne virus, one can comfortably conclude that the Wuhan coronavirus must very likely also come from nature and must share a recent common ancestor with RaTG13.
But here is the problem: this RaTG13 virus isn’t real. The evidence of its existence, its sequence, was fabricated.
Quite a claim, right? What is the claim based on? How can anyone fabricate a sequence? Who dares to carry out such a deceitful action and is not fearful of being caught? One is entitled to ask all of these questions. Now, let’s dig into each of them and see how the answers provided here may stand.
Who dares to carry out such a deceitful action?
The sequence of RaTG13 was reported by Zhengli Shi, a researcher from the Wuhan Institute of Virology and the biosafety level 4 (P4) lab for virology research. Dr. Shi is the top coronavirus expert in China. She has gained a nickname of “batwoman” because she and her team have a long history of capturing wild bats in caves all over for the purpose of detecting and sometimes isolating coronaviruses within them. As publicly stated, the goal of her research is to identify animal coronaviruses that have the potential of crossing-over to infect humans and thereby help the public avoid SARS-like disasters in the future.
Ironically, contrary to this self-portrait, since the very beginning of the current pandemic, Zhengli Shi has been singled out as THE suspect, who may have created the Wuhan coronavirus and, in doing so, caused a world-wide disaster. Interestingly, on Jan 23rd, 2020, just before this “rumor” started to soar though the roof, Shi published a paper in Nature (1), where she compared the freshly obtained sequence of the Wuhan coronavirus with those of other coronaviruses and thus delineated an evolutionary path of this new virus. In this publication, all of a sudden and out of nowhere, Shi reported this bat coronavirus, RaTG13, which pampered the public and seemingly helped shape a consensus in the field that the Wuhan coronavirus is of a natural origin.
As stated in the paper, RaTG13 was discovered from Yunnan province, China, in 2013. According to credible sources, Shi has admitted to several individuals in the field that she does not have a physical copy of this RaTG13 virus. Her lab allegedly collected some bat feces in 2013 and analyzed these samples for possible presence of coronaviruses based on genetic evidence. To put it into plainer words, she has no physical proof for the existence of this RaTG13 virus. She only has its sequence information, which is nothing but a string of letters alternating between A, T, G, and C.
Can the sequence of such a virus be fabricated? It cannot be any easier. It takes a person less than a day to TYPE such a sequence (less than 30,000 letters) in a word file. And it would be a thousand times easier if you already have a template that is about 96% identical to the one you are trying to create. Once the typing is finished, one can upload the sequence onto the public database. Contrary to general conception, such database does not really have a way to validate the authenticity or correctness of the uploaded sequence. It relies completely upon the scientists themselves – upon their honesty and consciences. Once uploaded and released, such sequence data becomes public and can be used legitimately in scientific analysis and publications.
Now, does this RaTG13 sequence qualify as credible evidence in judging the matter? Well, remember, a central part of the matter is whether or not this Wuhan coronavirus was engineered or created by ZHENGLI SHI. It is Shi, not anybody else, who is the biggest suspect of this possible crime that is grander than anything else in human history. Given the circumstances, wouldn’t she have a strong enough motive to be deceitful? If the evidence she raised to prove herself innocent was nothing but a bunch of letters recently typed in a word file, should anyone treat it as valid evidence?
RaTG13, if truly exists, should never be neglected by Shi for a period of seven years
Let’s now think about this from another direction. The sequence of RaTG13 is highly alarming – it clearly shows a potential of the virus to infect humans.
Within the spike protein of a β coronavirus, there is a critical piece named receptor-binding domain (RBD), which dictates whether or not this virus can use the ACE2 receptor on the surface of our cells and thereby infect humans. As a routine, when Shi’s team finishes collecting samples and confirms the presence of a coronavirus, the first thing they would do is to look at the sequence of the virus’ RBD. If there is resemblance between this sequence and that of the SARS virus (rarely so), their blood would boil because they have found something that may jump over to humans. It also means that top-journal publications are coming their way.
In 2013, Shi made her fame in the coronavirus field by publishing in Nature two bat coronaviruses (Rs3367 and SHC014), which share considerable sequence similarity with SARS in the RBD region (2). This work, for the first time, proved a bat origin of SARS. In the following years, her team continued to publish articles, featuring additional bat coronaviruses that share these important sequence motifs (3, 4).
What does an RBD sequence look like? Figure 1 is the sequence comparison between SARS RBD and the RBDs of the bat coronaviruses that Zhengli Shi published in high-profile journals (2-4). Comparing to SARS (top), many bat coronaviruses (most of the ones in the bottom half) had substantial deletions in their RBDs and are thus likely defective in targeting humans. In contrast, some bat coronaviruses (upper half) not only resemble SARS in the completeness of the RBD sequences but also contain amino acids similar to their SARS counterparts at some of the five locations known to be critical for binding human ACE2 receptor. This group of viruses, with these dazzling features, were perceived by the field as breakthroughs.

Figure 1. Sequence alignment comparing the RBDs of SARS (top) and RaTG13 (red arrow) to RBDs of bat coronaviruses that Zhengli Shi published in high-profile journals from 2013-2017 (2-4). Amino acid residues highlighted by Shi as critical for binding human ACE2 receptor (2) are labeled in red text on top. Alignment was done using the MultAlin webserver (http://multalin.toulouse.inra.fr/multalin/).
How does RaTG13, which was discovered in 2013, compare to these most cherished collections of Shi’s?
By appearance, RaTG13 clearly belongs to the “good-looking” group. It rivals with the best ones in its completeness of the RBD sequence as well as in the conservation of critical amino acids. While a single amino acid insertion is observed, it occurs in a variable region and can be easily tolerated without affecting the protein function. Importantly, RaTG13 preserves the binding motifs as much as, if not better than, any other bat coronavirus in Shi’s list. At position 442, RaTG13 has a “L”, which beats most, if not all, bat viruses in resembling the “Y” in SARS RBD (“L” and “Y” both mediate hydrophobic interactions). At position 472, RaTG13 is the only bat coronavirus that has the residue “L”, which is identical to SARS. Although the amino acids at the other three positions are not identical to their counterparts in SARS, they are all conservative mutations, which may not negatively impact the protein’s function. (In fact, a very recent publication confirmed that the RBD from RaTG13, like SARS RBD, can indeed bind the human ACE2 receptor (5). Note: the RaTG13 RBD gene used in this work was synthesized).
As an expert as Shi is, she only needed to take one peek at the sequence of RaTG13’s RBD and immediately realize: this virus closely resembles SARS in its RBD and has a clear potential of infecting humans. If Shi’s public statement is true and she indeed intends to discover bat coronaviruses with a potential to cross-over to humans, how could she possibly overlook this extremely interesting finding of RaTG13? If this RaTG13 was discovered SEVEN years ago in 2013, why did Shi not publish this astonishing finding earlier and yet let the “less-attractive” viruses take the stage? Why did she decide to publish such a sequence only when the current outbreak took place and people started questioning the origin of the Wuhan coronavirus?
None of these makes sense. These facts only add to the suspicion – Zhengli Shi either was directly involved in the creation of this virus/bioweapon, or helped cover it up, or both.
Of course, these facts also add to the claim that RaTG13 is a fake virus – it exists on Nature (the journal) but not in nature.
A closer look at the gene sequence of RaTG13’s spike reveals clear evidence of human manipulation
To assist our analysis here, we have to first understand one basic feature of natural evolution.
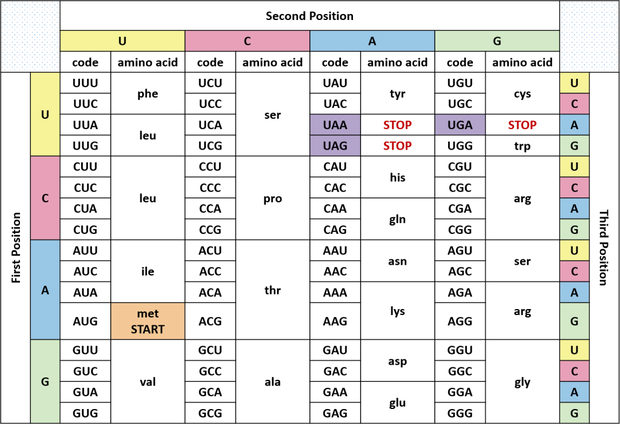
When a gene (composed of nucleotides) is being translated into a protein (composed of amino acids), every three consecutive nucleotides constitute a codon and each codon encodes a particular amino acid (Figure 2). On the other hand, an amino acid typically corresponds to four codons, although some amino acids have one or two more and some one or two less (you can learn more about this here: https://passel2.unl.edu/view/lesson/3ccee8500ac8/6). What does it mean? It means that, when a nucleotide has changed (or in other words a single nucleotide substitution has occurred), the codon is certainly altered but the corresponding amino acid may or may not change. This is because the new codon may encode the same amino acid as the old codon does. A single nucleotide substitution that results in NO change of the amino acid is referred to as a synonymous mutation. A single nucleotide substitution that leads to a change in amino acid is called a non-synonymous mutation. When evolution takes place through random mutations, on average, every six nucleotide changes result in the change of one amino acid. In other words, on average and under normal conditions, the ratio between the number of synonymous mutations and that of non-synonymous mutations should be around 5:1.
Now let’s further illustrate such a relationship using an example. In Figure 3A, synonymous and non-synonymous mutations are counted when the gene sequences of the spike proteins from two closely related bat coronaviruses, ZC45 and ZXC21 (6) are compared. The green curve depicts how the number of the synonymous mutation grows (Y axis) when the codons are analyzed sequentially (X axis). The red curve represents the trend of non-synonymous mutations. As expected, there are more synonymous mutations than non-synonymous mutations. Importantly, a correlation between the two curves is clearly present: they climb up and go through plateaus in a roughly synchronized manner. Throughout the whole length of the gene, at any point, the ratio between the accumulated synonymous and non-synonymous mutations is maintained at around 5:1. As we have described in the preceding paragraph, these features are consistent with what is the expected when two lineages closely relate to each other evolutionarily and the differences in their sequences are results of random mutations.
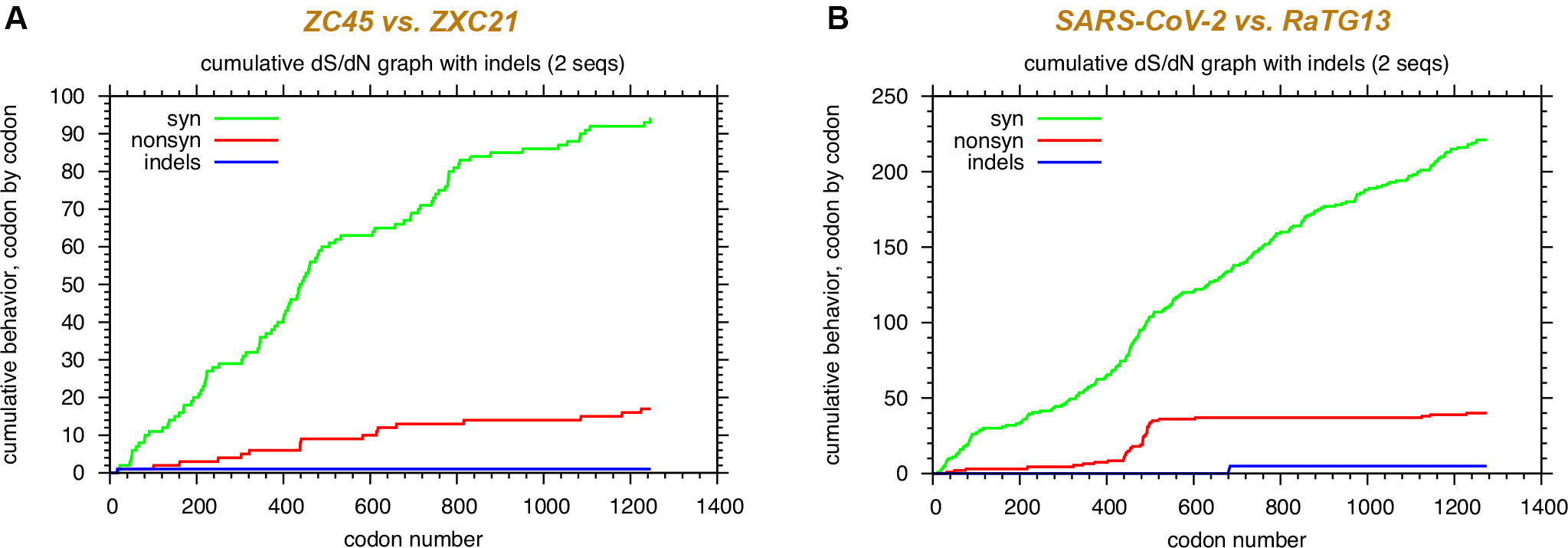
Does this hold true for RaTG13 and the Wuhan coronavirus? Not really.
Figure 3B is the same comparative analysis done between RaTG13 and the Wuhan coronavirus. One thing you can immediately appreciate is that, in the second half of the sequence, while the green curve continues to grow steadily, the red curve stays flat. For a region as wide as over 700 amino acids (corresponding to 2100 nucleotides), which is statistically substantial, the synchronization between the two curves is non-existent. Surprisingly, or maybe not so surprisingly, at the end, the final counts of synonymous and non-synonymous mutations yield a ratio of just over five, consistent with what’s expected out of natural evolution.
Let’s bring out some numbers to help us better comprehend the difference here. Let’s focus on the S2 protein, the second half of the spike ranging from 684 to 1273 (numbering according to the Wuhan coronavirus). Detailed analysis of this region reveals that, between ZC45 and ZXC21, a total of 32 nucleotides have changed and 5 of them lead to amino acid mutations (27 synonymous mutations vs. 5 non-synonymous mutations). It is, again, consistent with the scenario of natural evolution: every six nucleotide changes result in the change of one amino acid; synonymous/non-synonymous ratio is about 5:1. In contrast, for the same S2 region, between the Wuhan coronavirus and RaTG13, there are a total of 90 nucleotide changes and only two amino acid mutations. Here, every 45 nucleotide changes correspond to one amino acid change. The synonymous/non-synonymous ratio is 44:1.
It is noteworthy that ZC45 and ZXC21 share ~97% sequence identity, just like that between the Wuhan coronavirus and RaTG13. So, the above comparison is very proper and reliable.
If a person is studying the sequence differences between the Wuhan coronavirus and RaTG13 and yet pays attention only to the overall synonymous/non-synonymous ratio for the spike sequence, nothing would look strange. However, if one digs out as much details as shown in Figure 3, any person with a reasonable mind would say that something is clearly wrong.
What is the best way to interpret this? A safe conclusion is that, between the Wuhan coronavirus and RaTG13, at least one is non-natural. If one is natural, then the other one must be not. Of course, the other possibility also exists – neither of them came from nature.
If the Wuhan coronavirus is non-natural, then we have reached the end of our investigation.
In fact, the Wuhan coronavirus may “look” natural even if it is a bioweapon because it is most likely made by using a natural coronavirus as a template. This would lead to a conclusion that the RaTG13 is non-natural, which is consistent with the facts we have brought up earlier: no physical copies of the virus exist and the sequence is highly likely fabricated.
How could Zhengli Shi fail so badly in fabricating the RaTG13 sequence? While, when I said it was easy to type out a fake sequence that is 96% identical to a template, I did not say that it is easy to maintain a reasonable synonymous/non-synonymous ratio throughout the whole genome. Unfortunately for Shi, when she had to come up a good sequence for S1 and the RBD within it (she knows that this part will be scrutinized the most), she had somehow exhausted the number of non-synonymous mutations she could use here. To maintain a reasonable synonymous/non-synonymous ratio for the whole Spike-encoding gene (we can actually give her some credit here as she did remember to get it close to 5:1), she had to strictly limit the number of non-synonymous mutations in the S2 half of spike, which ended up flattening the red curve. It is hard to be a cheater after all.
A deeper reason that Shi and the CCP needs the cover of RaTG13
Hopefully you are now as convinced as I am in that the RaTG13 sequence is indeed a fabrication. The first thing we should do then is to discredit any scientific publication, which based its analysis on the RaTG13 sequence and subsequently arrived at the conclusion that the Wuhan coronavirus is of natural origin. When you do a clean-up like that, you will see that there is practically nothing left.
Next, we can look back to see what other coronaviruses are close to the Wuhan coronavirus in terms of sequence similarity. It turns out the two bat viruses featured in Figure 3A, ZC45 and ZXC21, are the next hits, each sharing 95% amino acid sequence identity (~89% nucleotide identity) with the Wuhan coronavirus. What is striking is the manner that the Wuhan coronavirus resembles these two bat coronaviruses – while every other protein remains highly identical, the S1 part of Spike, which dictates host selection, is only 69% identical. I have posted an article earlier, where I thoroughly analyzed this pattern and discussed how it is interlocked with the Wuhan coronavirus being a bioweapon made with ZC45 or ZXC21 as a template (www.nerdhaspower.weebly.com).
One thing we haven’t mentioned so far is that ZC45 and ZXC21 are bat coronaviruses discovered, collected, and published by a military research lab of the Chinese Communist Party (CCP) (6). They are owned only by the CCP. Now you may be able to appreciate the full benefits that the CCP creates by reporting a fake RaTG13 virus with a fabricated sequence – it would just be too obvious otherwise.
Finally, I would like to add an additional piece of evidence, which was brought up in a comment of my earlier article by someone who is clearly an expert. It, in my opinion, hugely strengthens the claim that the Wuhan coronavirus is of non-natural origin.
The E protein of β coronaviruses is a structural protein that is tolerant of mutations as evidenced both in SARS and in bat coronaviruses. However, on the amino acid level, E protein of the Wuhan coronavirus identified at the beginning of the outbreak is 100% identical to those of the suspected templates, ZC45 and ZXC21 (Figure 4). What is striking is that, after a short two-months spread of the virus in humans, the E protein is already mutating. Sequence data obtained within the month of April indicate that mutations have occurred to four different locations (Figure 4). Note that the E protein makes very limited interactions with host proteins and thus is not under evolutionary pressure to adapt to a new host. Not only the E protein can tolerate mutations but also its mutational rate is held constant across different coronavirus species. The fact that the E protein of the Wuhan coronaviruses is already mutating in the short period of human-to-human transmission is consistent with its evolutionary feature. In stark contrast, while ZC45/ZXC21 and the Wuhan coronavirus are more distant evolutionarily, the E proteins within them are 100% identical. In no way this could be a result of natural evolution.
The most plausible explanation: the Wuhan coronavirus is a bioweapon made using ZC45/ZXC21 as a template.
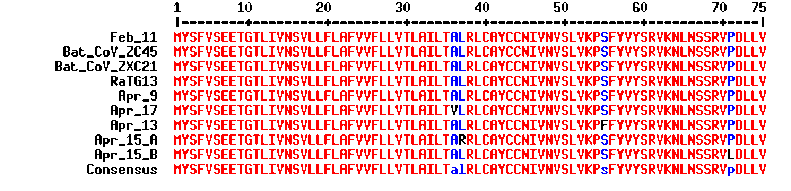
Following the suggestion given by Viennah K. Erchus, additional information is added here to illustrate how E proteins are tolerant of mutations. As shown in Figure 5, mutations and insertion/deletions in E proteins have been observed at multiple locations both in SARS coronaviruses and in bat coronaviruses. This clearly indicates E protein’s tendency and permissiveness toward mutations across β coronavirus species. What is inconsistent with this trait is the fact that ZC45/ZXC21 and the Wuhan coronavirus, while significantly distant from each other in evolution, share 100% identity in E proteins. Again, in no way this could be a result of natural evolution. This further supports the claim that the Wuhan coronavirus is made in a lab by following ZC45/ZXC21 as a template.
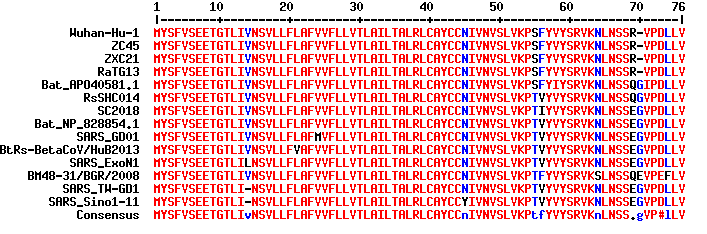
Figure 5. Sequence alignment of E proteins from Wuhan coronavirus (Wuhan-Hu-1), SARS coronaviruses (SARS_GD01, SARS_ExoN1, SARS_TW_GD1, SARS_Sino1_11), and bat coronaviruses (Bat_AP040581.1, RsSHC014, SC2018, Bat_NP_828854.1, BtRs-BetaCoV/HuB2013, BM48-31/BGR/2008). The ready-for-analysis sequences were kindly prepared by Viennah K. Erchus.
Author’s note and acknowledgement:
The current writing is a reproduction of part of my earlier article (www.nerdhaspower.weebly.com), with two significant additions. The first addition is the analysis of synonymous/non-synonymous mutations of Spike-encoding gene of RaTG13. This was first identified and analyzed by Elannor D. Allens, who described this finding in a comment under my earlier article. Using the synonymous/nonsynonymous ratio between ZC45 and ZXC21 as an example of natural evolution was suggested by 冠军的亲爹. The second addition is the analysis of recently observed mutations in the viral E proteins. This was first observed and analyzed by John F. Signus, who also posted his findings in a series of comments. The writing here owes greatly to their brilliance and insights. 冠军的亲爹 particularly encouraged me in pursuing this writing. Although these names may only be their identities online, I nonetheless feel that it is appropriate to give the credits using these names. I believe dearly that the persons behind these names are the most decent and brilliant kind.
* Just realized that I used the wrong format for this page. If you want to leave a comment, please go to the main page “BLOG”. Sorry for the inconvenience.
References:
1. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020.
2. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503(7477):535-8.
3. Zeng LP, Gao YT, Ge XY, Zhang Q, Peng C, Yang XL, et al. Bat Severe Acute Respiratory Syndrome-Like Coronavirus WIV1 Encodes an Extra Accessory Protein, ORFX, Involved in Modulation of the Host Immune Response. J Virol. 2016;90(14):6573-82.
4. Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13(11):e1006698.
5. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020.
6. Hu D, Zhu C, Ai L, He T, Wang Y, Ye F, et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg Microbes Infect. 2018;7(1):154.
7. Korber B. HIV Signature and Sequence Variation Analysis. Computational Analysis of HIV Molecular Sequences. 2000;Chapter 4:55-72.



